Role of Nitric Oxide Signaling Pathway in the Cytosolic and Mitochondrial Signaling of IPC
It was reported that when the isolated rabbit heart was perfused with N (G)-nitro-L-arginine methyl ester (L-NAME), the non-specific NOS inhibitor, at 10 min before the coronary occlusion and continued for 15 min of reperfusion period in 45 min of ischemia and 180 min of reperfusion episode, the infarct size was decreased compared to the untreated group, in addition to that, the NOS inhibitor could reduce infarct size in non-preconditioned hearts after I/R injury. Also discovering the important role of iNOS in the cardioprotective effect against I/R injury when aminoguanidine (AMG), the specific-iNOS inhibitor, was used. In groups treated with AMG alone or with MLA, the infarct size was significantly increased compared to the MLA-only treated group and was not different from the vehicle-treated group. These results suggested that when iNOS is inhibited, the cardioprotective effect of MLA disappears. In a pacing-induced preconditioning model of the rat heart, NG-nitro-L-arginine (L-NNA), a non-specific NOS inhibitor, increased the release of lactate dehydrogenase, a marker of necrotic cell death, from the coronary-occluded ischemic area. L-NAME could abolish the cardioprotective effect of adenosine receptor agonist 5’-(N-ethylcarboxamido) adenosine (NECA) or bradykinin in the I/R model in rabbit hearts. Prendes et al. demonstrated in 2007 that IPC increased cardiac contractility after global ischemia and reperfusion in isolated rat hearts; however, the effect of IPC disappeared when treated with L-NAME. Another study done by Nakano et al showed that S-nitroso-N-acetylpenicillamine (SNAP), an NO donor that serves as the resource of exogenous NO, was able to mimic preconditioning by decreasing infarct size in rabbit hearts after a long period of I/R without IPC, compared to those without SNAP infusion. It was discovered that IPC, with a sequence of 6 cycles of 4-minute coronary occlusion and 4-minute reperfusion, increased the expressions of iNOS mRNA and protein at 3 hours after the last cycle of IPC; however, the expression of eNOS remained unchanged. Guo et al. found that iNOS knockout mice had larger infarct size than wild-type mice after being subjected to 30-min coronary occlusion and 24-h reperfusion in the presence of IPC (6 episodes of 4-min occlusion and 4-min reperfusion cycles). Moreover, at 24 h after I/R, wild-type mice with IPC had smaller infarct size than both non-IPC wild-type and iNOS knockout mice at 24 h after IPC. This cardioprotective effect of iNOS was supported by the increased level of iNOS expression in the IPC group compared to the non-IPC group at 24 h after IPC. In eNOS knockout mice, these findings suggested that the role of eNOS in preconditioning is less prominent than that of iNOS. Although the infarct size was reduced in the overexpressed eNOS compared to wild type mice, du Toit et al. found that there was no difference in the infarct size between IPC and non-IPC in this overexpressed eNOS mouse model. A growing evidence demonstrates the important relationship among NO, ROS, and ischemic preconditioning. In 2003 Lebuffe et al. found that H2O2 and NO were important for preconditioning-like cardioprotection. Furthermore, when the mitochondrial ATP-sensitive K+ (mitoKATP) channel was inhibited by 5-hydroxydecanoate (5-HD) in the IPC model, the cardioprotective effect of IPC disappeared, suggesting the importance of the mitoKATP channel in cardioprotection against I/R injury. Recent research on the mechanisms of IPC suggests that during a brief episode of ischemia, 3 ligands are released from the cardiomyocytes, and long sequences of activities are found here. These ligands (bradykinin, endogenous opioid, and adenosine), occupying their respective G-protein coupled receptors (GPCRs), result in activations of phosphatidylinositol 3-kinase (PI3K) and series of phospholipid-dependent kinase (PDK). PDK causes phosphorylation and activation of Akt, where the latter induces further phosphorylation onto NOS, causing NO to be generated. After that, sGC, activated by NO, transforms GTP into cGMP, in which PKG is finally activated. In the last step of cytosolic signaling, PKG then reacts on mitochondria, resulting in the opening of the mitoKATP channel, and the opening of the mitoKATP channel leads to the inhibition of the mitochondrial permeability transition pore (mPTP), resulting in the protection of mitochondria from damage during ischemia. Thus, a cGMP-dependent mechanism has been proposed as the main pathway to activate the mitoKATP channel via phosphorylation by PKG (Fig. 15).
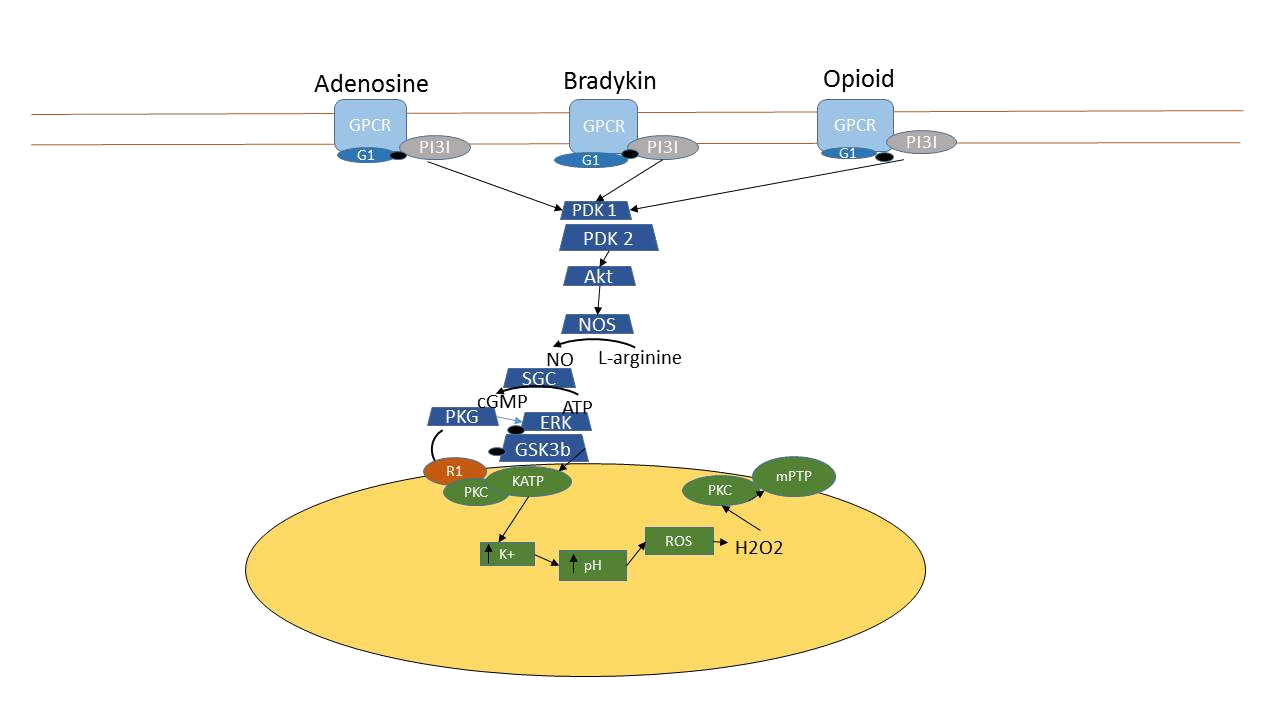
Figure 15. The schematic diagram represents the ischemic preconditioning pathway in cardiomyocytes. IPC induces cardiomyocytes to release adenosine, bradykinin and endogenous opioid which occupy their specifi G-protein coupled receptors. After that, the signal will pass into the cytosol via the activation of the enzyme series including PI3K, PDKs, Akt and NOS. The latter enzyme produces NO which acts as signaling molecule to activate sGC and resulted in the cGMP formation. Then, cGMP activates PKG which has the separated mechanism on the mitochondria, the direct and indirect mechanism. The direct mechanism of PKG is on the R1 protein on outer mitochondrial membrane which then activates the opening of mitoKATP channel on the inner mitochondrial membrane via the phosphorylation of PKCε1. After the mitoK channel opening, K+ then enters the ATP mitochondrial matrix and H+ is ejected out of the matrix to balance the positive charge. When H + level decrease, the electron transport chain is interrupted and leads to the formation of superoxide anion and then H2O2. Finally, H2O2 will act as signaling molecule to activate PKCε2 which then inhibits the opening of mitochondrial permeable transition pore (mPTP). This inhibition of mPTP opening helps to protect mitochondrial damage during I/R injury