Complexities of coupling NOS pathways in the heart
The 3 nitric oxide synthase (NOS) isoforms have been found in mammalian heart tissues. The endothelial isoform of NOS (eNOS, or NOS3) robustly expressed in the endothelial cells of the cardiac vasculature, and also present within cardiac myocytes, where the enzyme associates with the scaffolding/regulatory protein caveolin-3 in T tubules in plasmalemmal caveolae. Furthermore the neuronal NOS (nNOS, or NOS1) is also expressed in cardiac myocytes, where the enzyme appears to be localized in the sarcoplasmic reticulum and modulates phospholamban phosphorylation. Though both eNOS and nNOS appear to be physiologically expressed in cardiac tissues, the inflammation-related NOS isoform (iNOS, or NOS2) only appears in the heart after immunoactivation; iNOS may modulate the decline in myocardial function seen in sepsis. A study by Silberman et al. brings some new insights into NOS pathways in the heart. Mouse model that develops diastolic dysfunction in the absence of systolic dysfunction or cardiac hypertrophy, and cardiac tissues from these mice showed increased oxidation of a key NOS cofactor, tetrahydrobiopterin (BH4), as well as increased superoxide formation. It was shown later that cardiac NOS had been “uncoupled” in this model of diastolic dysfunction, and they show that BH4 feeding promoted the restoration of “coupled” eNOS and normalized diastolic function. All 3 NOS isoforms catalyze the formation of NO by oxidizing the amino acid L-arginine to form NO plus L-citrulline. NAPDH and molecular O2 serve as obligate cosubstrates, in addition to the ubiquitous calcium-binding signaling protein calmodulin which is an essential allosteric activator that binds to and activates NOS. Also several redox-active cofactors are required for NOS catalysis, including: FAD (flavin adenine dinucleotide), FMN (flavin mononucleotide), heme, and BH4. Under different conditions, all 3 NOS isoforms also have the capacity to synthesize ROS such as superoxide (O2-) instead of their usual product, NO. The NOS-dependent O2- formation was first described in studies of purified NOS proteins and was termed NOS “uncoupling” because the usual catalytic electron flow within the enzyme is uncoupled from NO synthesis and is instead diverted to molecular oxygen to yield O2- Therefore purified NOS can produce O2- when the key NOS cofactor BH4 is oxidized to form dihydrobiopterin (BH2); importantly, O2- production from purified NOS is not substantively affected by the NOS substrate arginine or by most arginine-based NOS inhibitors. Still it is not clear that O2- formation in cells containing NOS perforce represents NOS uncoupling; however there are many intracellular sources of O2- and extracellular determinants of redox homeostasis. The increase in O2 formation in cells or tissues in which NOS isoforms also happen to be present does not necessarily mean that the formation of superoxide is a direct consequence of NOS catalysis gone away.
In most mammalian tissues the major source for ROS is not “uncoupled” NOS; rather, ROS principally derived from mitochondria (Fig. 13) as a consequence of normal cellular oxidative metabolism.
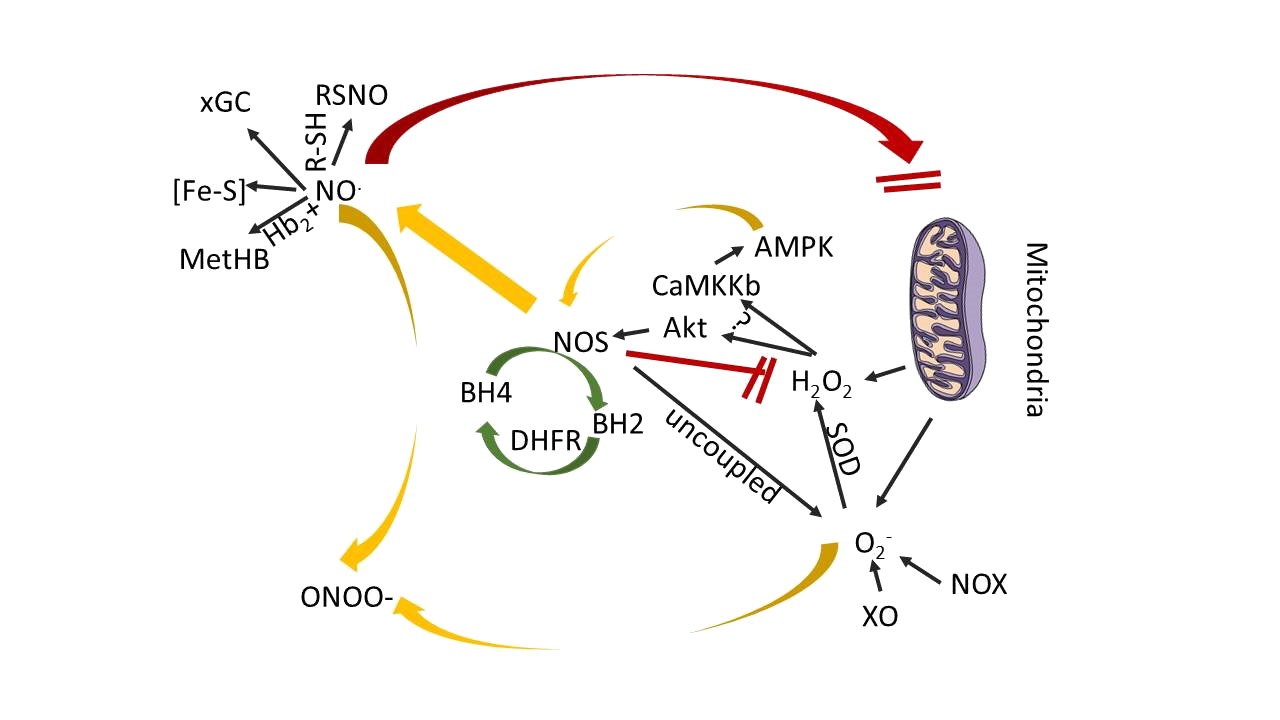
Figure 13: Scheme showing the principal reactions of NOS-derived products in cardiovascular tissues
Molecular oxygen is, of course, the ultimate acceptor of the electrons that flow through mitochondria, yielding H2O as a final product. But along the way, a 1-electron reduction of O2- can yield (sequentially) the ROS superoxide (O2-), hydrogen peroxide (H2O2), and the highly unstable hydroxyl radical (OH) before H2O is finally formed. H2O2 is the most abundant and stable of these ROS in most mammalian cells, and is a key signaling molecule that promotes eNOS activation in endothelial cells. O2 is rapidly reduced by superoxide dismutase to form H2O2, but O2 can also rapidly interact with NO to form peroxynitrite, which itself is a reactive oxidant molecule with important cellular effects. On the other hand, NO can also interact directly with mitochondrial electron transport proteins, and under some conditions, it may influence cellular respiration and ATP synthesis in the heart. The isoform eNOS appears to play a role in mitochondrial biogenesis, and NO adducts appear to modulate cardiac mitochondrial respiration. According to recent studies eNOS was reported to exert a tonic inhibitory effect on H2O2 synthesis in cultured endothelial cells. That study reported an increase in endothelial cell H2O2 synthesis after NOS enzyme inhibition, a situation sometimes ascribed to NOS “uncoupling.”
The increase in H2O2 synthesis after eNOS knockdown was blocked by small interfering RNA–mediated knockdown of the AMP-activated protein kinase and related proteins in the AMP-activated protein kinase pathway, which implicates cellular phosphorylation pathways in at least some of the NOS-related changes in ROS formation (Fig. 13). Also NOS uncoupling can be influenced by factors independent of BH4, for example, post-translational modifications may play a role in NOS-dependent O2 synthesis: Chen and colleagues have shown that phosphorylation of eNOS at serine 1177 increases O2 synthesis by the enzyme, whereas eNOS phosphorylation at threonine 495 does not. Several of the studies that have explored the pathways whereby BH4 ameliorates diastolic dysfunction or endothelial dysfunction have provided evidence that implicates the renin-angiotensin system in the pathophysiological response. Angiotensin II modulates ROS production in diverse cardiovascular tissues and has been shown to stimulate eNOS derived O2- production, which in turn is suppressed by BH4. Also it was shown previously that the effects of BH4 were due to effects on NOS pathways in cardiac myocytes but did not involve activation of cGMP-dependent pathways in the heart.