Endothelial protection and signaling
NO is a reactive molecule that can rapidly diffuse throughout the cell. It can react with sulfur groups on cysteine residues in proteins, particularly when these cysteines are present in an acid–cysteine–base motif. The later results in reversible S-nitrosylation of these proteins and in the presence of reducing enzymes, such as GSNO-reductase, may also lead to disulphide bridges between cysteine residues in proteins. Furthermore, because of its unpaired electron it can associate with transition metals such as heme leading to nitrosyl modifications. Interestingly, eNOS is not a prerequisite for life and genetic deletion of eNOS appears to have little effect on basal vasodilation or basal leukocyte adhesion to the endothelium, supporting a more permissive role for NO under basal conditions. If one reviews the most important cellular effects of NO they seem to be involved in conservation of metabolism. Probably one important function of NO in the (endothelial) cell is its competition with oxygen as an electron acceptor at complex IV in oxidative phosphorylation. This means that with diminishing oxygen availability NO will predominate as an electron acceptor and reduce oxidative phosphorylation thus limiting energy expenditure. NO also participates in defense against hypoxia by increasing hypoxia-inducible factor-1(HIF-1) binding activity and HIF-1protein levels. A very interesting feature of the NOS enzymes is that they not only generate NO but may also produce reactive oxygen species themselves. Basically, eNOS, as well as the other NOS isoforms, are NADPH consuming enzymes. As a consequence there will be electrons flowing in the enzyme from the reductase domain toward the heme containing oxygenase domain.
In the eNOS enzyme, the cofactor calmodulin is an important checkpoint in this chemistry as it is required for shuttling of electrons toward the heme group. The eNOS cofactor tetrahydrobiopterin (BH4), another important checkpoint in the chemistry, facilitates the reaction of electrons and oxygen with L-arginine leading to the formation of L-citrulline and NO. In the presence of oxygen a very reactive superoxy ferrous–peroxy ferric complex is formed, and if either BH4 or the substrate L-arginine are lacking, the superoxy ferrous–peroxy ferric complex may dissociate to yield superoxide (if BH4 is lacking) or H2O2 (if L-arginine is lacking). This latter state is referred to as the so-called “uncoupled” state of eNOS. As explained before, uncoupling of eNOS implies that the endothelial cell switches from a quiescent state (NO) into a state that is adapted for host defense (H2O2). In this respect generation of reactive oxygen species by uncoupled eNOS could be regarded as physiological signaling during injury and infection, and in fact could even be an essential effector mechanism in the host defense response, and eNOS uncoupling may represent a widely spread physiological mechanism. As vascular inflammation involves endothelial cell activation as well as concomitant influx of leukocytes, both uncoupling of eNOS as well as iNOS induction in leukocytes could modulate this immune response. The role of iNOS induction in host defense is well recognized. Recently, however, it has been suggested that iNOS induction may in fact be secondary to eNOS uncoupling. For example, during endotoxemia a marked reduction in inducible NO synthase protein was observed in the heart of eNOS knock-out mice compared with wild type, indicating that for iNOS induction initial eNOS activation is required. If uncoupling of eNOS is involved in host defense one would also expect increased susceptibility to infection in the absence of the eNOS enzyme. eNOS enzyme should have a Janus face, which allows the enzyme to switch from the coupled to the uncoupled mode when it encounters infectious agents from cardiovascular homeostasis to host defense.
Activation and endothelial deposition of the complement cascade, a first-line bacterial defense component of the innate immune system, causes redox signaling in the endothelial cell. Also, phagocytic cells take up bacteria through toll-like receptors or Fcy receptors and activate NADPH oxidase. Such activation of NADPH oxidases in phagocytes has been shown to induce downstream redox signaling in endothelial cells when these phagocytes adhere to endothelium. Many cell types including endothelial cells, furthermore, produce low levels of H2O2 in response to cytokines (transforming growth factor [TGF]-β tumor necrosis factor [TNF]-α, and interleukin [IL]), peptide growth factors (PDGF; EGF, VEGF, bFGF), and agonists of G protein–coupled receptors (eg: angiotensin II, thrombin, lysophosphatidicacid, sphingosine 1-phosphate, histamine, and bradykinin) involved in inflammation. A very important feature of BH4 is that it is very susceptible to oxidation and in fact can auto-oxidize leading to accelerated degradation of BH4 under conditions of oxidative stress and redox signaling. In this way activation of the innate immune system will also induce uncoupling of eNOS and subsequent proinflammatory transcription in the endothelial cell (Fig.14).
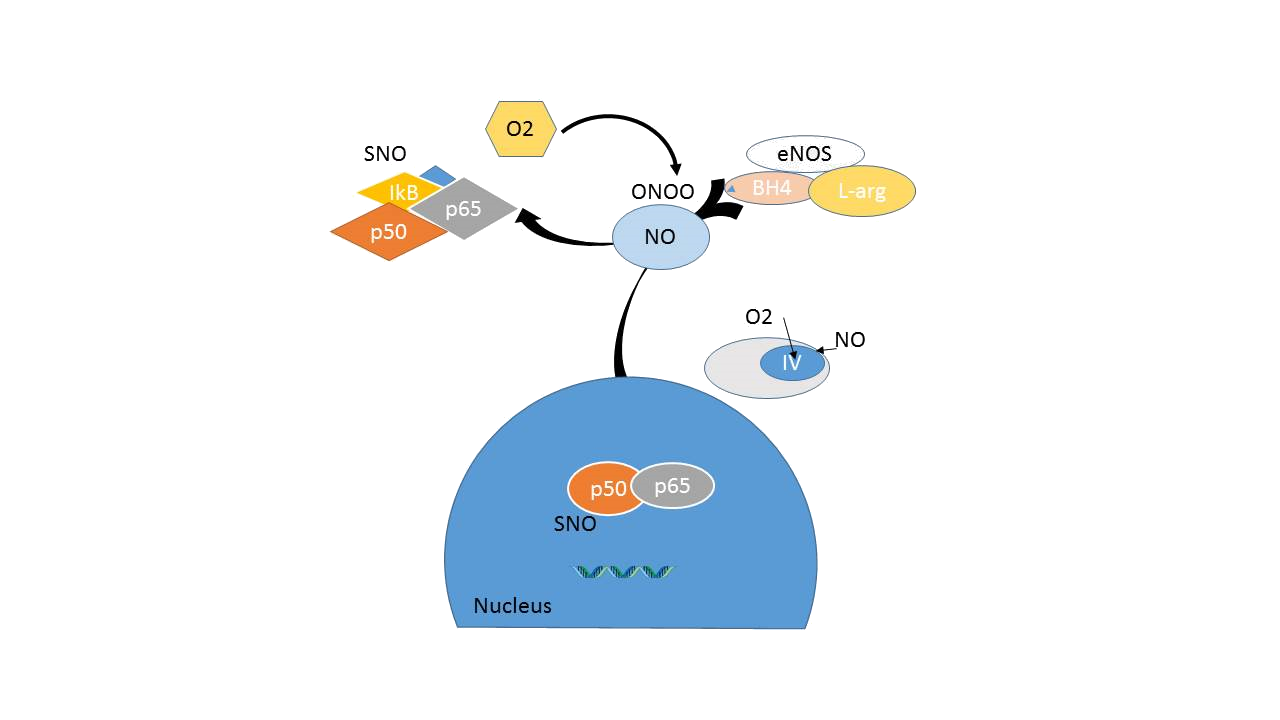
Figure 14A : esting endothelial function. In the presence of sufficient amount of tetrahydrobiopterin (BH4) and the substrate L-arginine, the eNOS enzyme produces NO. S-nitrosylation of transcription factors involved in endothelial activation such as NF-ƙB and cell-cycle proteins, the endothelial cell maintains a quiescent state. In addition, NO functions in the mitochondria to limit energy expenditure.
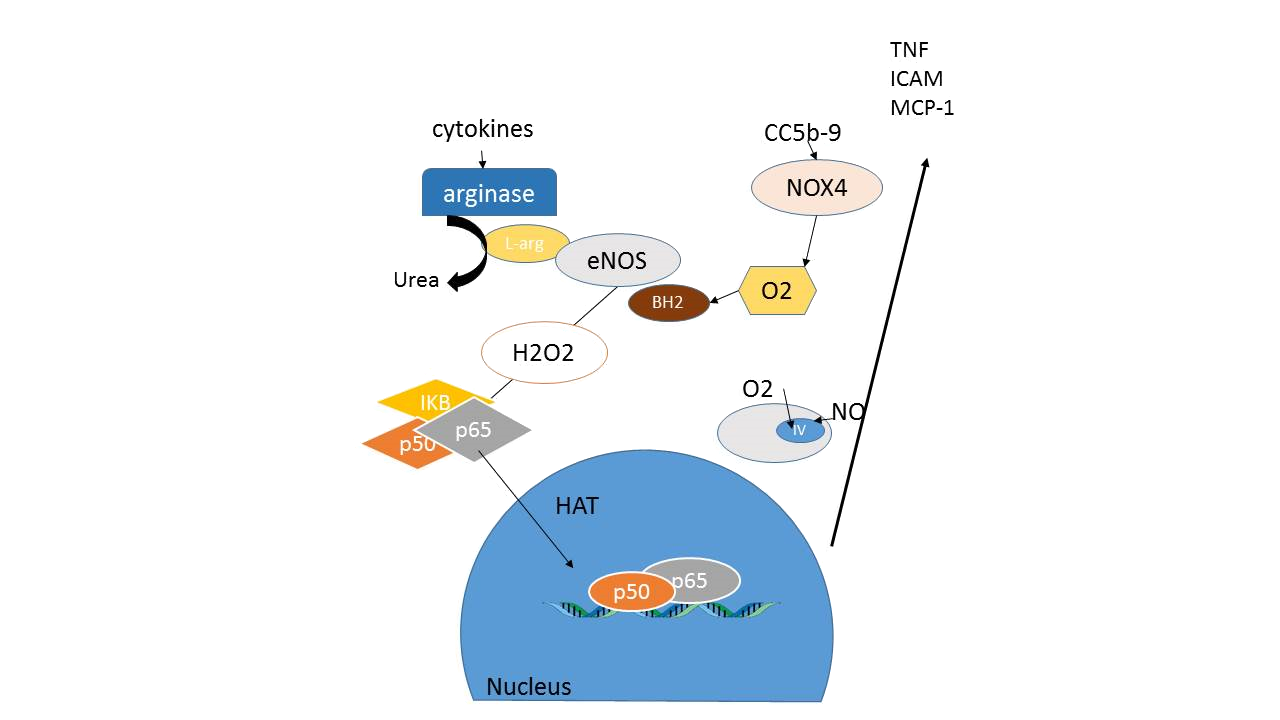
14B: Endothelial activation. In the presence of cytokines or complement activation the endothelial isoform of NADPH oxidase (NOX4) is activated leading to oxidative stress and oxidation of the cofactor tetrahydrobiopterin into BH2. In addition, arginase is induced shuttling the eNOS substrate L-arginine toward L-ornitine and urea production. As a result eNOS produces oxyradical species leading to H2O2 signaling into the cell.
Cytokines that are released by phagocytes will not only reduce BH4 availability, they can also induce arginase in the endothelium and shuttle L-arginine metabolism toward urea production. This may reduce L-arginine availability for the eNOS enzyme and further contribute to eNOS uncoupling. L-arginine deficiency at the eNOS enzyme may also result from increased presence of endogenous inhibitors of NOS such as ADMA and L-NMMA.