Blood pressure regulation
Short-term blood pressure regulation: Fast acting blood pressure control mechanisms include the baroreceptor reflexes, chemoreceptor reflexes, the adrenal medullary mechanism and the central nervous system ischemic response.
Baroreceptor Reflexes: As previously noted, these are stretch-sensitive sensory receptors located along the walls of most of the large arteries and are more abundant in the area of the carotid sinus at the base of the internal carotid artery and in the walls of the aortic arch. These reflexes are short-term and rapid acting, and do not cause major global changes in blood pressure. The baroreceptors eventually adapt to elevated blood pressures and will not decrease blood pressure to baseline (pre-stretch activation) values.
Adrenal medullary mechanism: This fast-acting mechanism is activated under various conditions such as large decreases in blood pressure, sudden and substantial increases in physical activity and other stressful conditions.
Chemoreceptor reflexes: These processes are activated when oxygen levels in the blood decrease, carbon dioxide levels increase and pH increases according to mechanisms of action discussed previously in this document.
Central nervous system (CNS) ischemic response: This mechanism under normal conditions does not play an important role in regulating blood pressure. CNS signaling elevates blood pressure in response to a lack of blood flow to the medulla oblongata of the brain.
Long-term blood pressure regulation: The major long-term regulatory mechanisms include the renin-angiotensin-aldosterone system, vasopressin (ADH), fluid shift, atrial natriuretic and stress-relaxation response mechanisms.
Renin-angiotensin-aldosterone system: These processes are important components of the endocrine system for control of blood pressure. Stimulation by reduced salt intake or sympathetic nervous system signaling, causes release of renin from the kidney and more precise the juxtaglomerular apparatus. Renin converts angiotensinogen to angiotensin I, which is converted to angiotensin II in the lungs by ACE (angiotensin converting enzyme). Angiotensin II is a potent vasoconstrictor. It causes an increase in blood pressure by increasing the total peripheral resistance and venous return to the heart. Angiotensin II stimulates the release of aldosterone from the adrenal gland which causes even more increase of blood pressure by promoting sodium and water retention, therefore an increase in blood volume. Angiotensin II also increases ADH secretion, salt appetite and thirst.
Vasopressin (ADH): ADH is released from the pituitary in response to changes in blood pressure that are detected by baroreceptors in the organ. ADH acts on blood vessels causing vasoconstriction, decreasing the rate of urine production by the kidneys, thereby helping maintain blood volume and blood pressure. ADH is also released from the pituitary in response to increase of solutes concentration in plasma, which can happen during dehydration or accidents involving plasma loss.
Atrial Natriuretic Factor: A hormone called atrial natriuretic factor is released from cells in the atria of the heart in response to stretching of the atrial cardiac muscle cells due to increased venous return. This hormone increases urine production by the kidneys, promoting loss of water and Na+, thereby decreasing blood volume, in turn decreasing venous return. It also causes vasodilation of arteries and veins thereby reducing total peripheral resistance. These two aforementioned modes of action by atrial natriuretic hormone reduce blood pressure. The three mechanisms mentioned above work together to regulate blood pressure by controlling urine production through the kidneys.
Fluid shift: This mechanism is activated in response to small changes in pressure across capillary walls. When blood pressure rises, fluid from the capillaries is forced into the interstitial spaces. This fluid movement causes reduction in blood volume and as a consequence the blood pressure decreases.
Stress-Relaxation response: This is a process by which smooth muscle cells of the blood vessels walls relax and contract. When blood volume increases rapidly, blood pressure increases and the smooth muscle cells relax, resulting in a more gradual increase in blood pressure. The opposite occurs when blood volume decreases rapidly: blood pressure decreases, smooth muscle cells contract, thereby reducing the volume of the blood vessels and counteracting further decline in blood pressure.
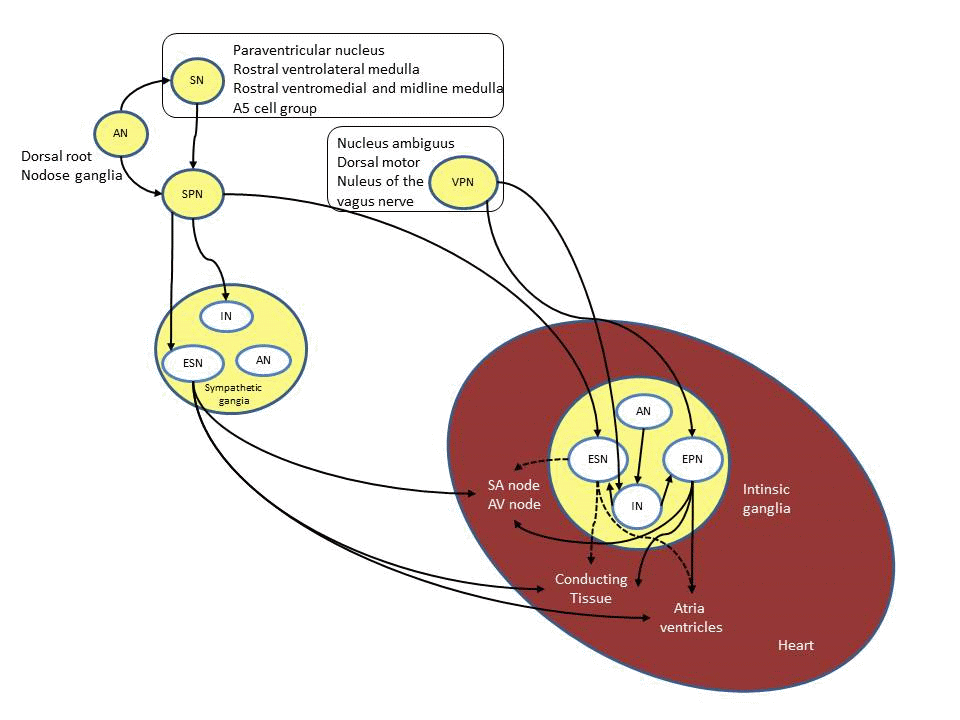
Figure 1. Schematic representation of the regulation of the blood pressure
Electrophysiology of the heart: The initial event of a cardiac contraction, or a heartbeat, is cardiac depolarization. Normally, the initial depolarizing action potential (AP) stimulus originates in the sinoatrial (SA) node, located in the posterior wall of the right atrium at its junction with the superior vena cava. The SA node is a cluster of pacemaker cells which exhibit automaticity, meaning that they fire APs spontaneously without any external stimuli. The electrical impulse then spreads through the entire atrial muscle mass and eventually to the atrioventricular (AV) node, which in the normal heart is the only electrical connection between the atria and ventricles. On an electrocardiogram (ECG) the time it takes the electrical impulse to travel from the SA node through the AV node is represented by the PR interval. It is important that an electrical impulse is delayed to allow the atria to finish its contraction before the ventricles begin theirs. It is mainly the AV node that is responsible for this delay and is mostly due to diminished number of gap junction between cells in this area. After conduction through the AV node the electrical impulse travel down the His-Purkinje system and then to the ventricular muscles and causing a contraction.
Action potential mechanisms: AP occurs when the membrane potential of a cell depolarizes suddenly and then returns to its resting state. Two types of AP exist in the heart, the non-pacemaker AP of the myocardial contractile cells and the pacemaker AP of myocardial autorhytmic cells like the once found in the SA node.
Non-pacemaker AP: This form of AP may be divided into 5 phases.
Phase 1 represents the initial phase of repolarization and is caused by efflux of K+ through the transient outward K+ channel (Ito), during which time Ina are inactivated. L-type calcium (Ca2+) channels that started opened at around -40mV and cause an inward Ca2+ movement that is are responsible for phase 2 or the plateau phase. Phase 3 represents rapid repolarization, during which L-type Ca2+ channels close and K+ channels called delayed rectifier (IKr) open and repolarize the cell back to its resting membrane potential and a new cycle can begin (Klaubunde 2004). Phase 4 represents the initial phase, in which the cell is at its resting membrane potential (Em) of around -90mV. The Em of a cell is determined mainly by the concentration gradients of individual ions and the permeability of the membrane to these ions. At resting state the cell membrane has a finite permeability to potassium ions ( K+) but very little permeability to sodium ions (Na+) resulting in the Em close to the equilibrium potential for K+ (EK). The ion channel responsible for a constant efflux of K+ and maintaining steady Em is called the inward rectifier (IK1). Sudden depolarization, e.g. due to a travelling wave from an adjacent cell, to a threshold of about -70 mV, results in the opening of fast Na+ channels (INa) along with decrease in K+ conductance. This results in a rapid AP upstroke to around +20 mV, representing phase 0 of the AP.
Pacemaker AP: Pacemaker cells are unique in a way that they are able to generate their own AP spontaneously. The underlying mechanism for their automaticity lies in the fact that they do not have a true resting potential like non-pacemaker cells, they lack the inward rectifier (IK1). Instead they have an unstable membrane potential, which after each AP starts at -65 mV and then slowly drifts up to threshold at around -40 mV, this represents the phase 4 of the pacemaker AP. Unlike in non-pacemaker AP, the early repolarization (phase 1) and the plateau phase (phase 2) do not exist. There are two main ion channels responsible for this unstable membrane potential. The first one is a slow Na+ channel (If) also called pacemaker or “funny current” and the second one is T-type calcium channel. When the unstable membrane potential reaches a threshold of around -40mV L-type calcium channels open and depolarize the cell, which represents phase 0 of the AP. These are slow calcium channels causing phase 0 to be much slower in pacemaker cells compared to non-pacemaker cells. After depolarization, the delayed rectifier potassium channels (IKr) open and the L-type calcium channels close, this repolarize the cell and represents phase 3. As the cell repolarizes, potassium channels closes, If channels open and phase 4 begins. If channels are responsible for the initial depolarization of phase 4, they are permeable to both Na+ and K+, but Na+ influx exceed K+ resulting in a net influx of positive charge, thus slowly depolarizing the cell. In the second half of phase 4, T-type Ca2+ channels begins to open further depolarizing the cell and finally L-type Ca2+ channel open when the cell reaches the threshold.
Refractory period and post-depolarization: During phases 0, 1, 2 and early phase 3, non-pacemaker cells are unexcitable. This phase of pacemaker function is called the effective refractory period (ERP). This is a latency interval, in which pacemaker activity remains inactive. It serves as a protective mechanism for the heart, by inhibiting new AP and thus new contractions from occurring before the previous one has completed its cycle. This critical coordinating function prevents tetany and allows sufficient time for diastolic filling of the heart. The ERP also blocks occurrence of reentry arrhythmias by preventing newly generated AP from travelling in the opposite directions. Two factors play key roles in the mechanism of long ERPs typical of cardiac cells (by contrast with those in skeletal muscle cells). One contributor is the AP duration (APD) which is much longer in cardiac myocytes versus skeletal muscle APDs, primarily due to length of the plateau phase. The second factor is a consequence of the activity of voltage gated channels, including fast sodium channels. Fast sodium channels have two gates, an activation gate (m-gate) and an inactivation gate (h-gate). At resting membrane potential, the m-gate is closed and h-gate open. When the cell reaches threshold, the m-gate opens and the h-gate begins to close. Significantly, the m-gate opens more rapidly than the h-gate closes allowing Na+ influx and depolarization of the cell. The h-gate will remain closed until the end of repolarization when it opens and the m-gate closes, making the cell ready for the next cycle.
During ERP the cell is still repolarizing and the gates of the sodium channels have not returned to their resting state making it impossible to initiate a new AP. A relative refractory period (RRP), follows the ERP at a point where a supra-threshold is able to evoke AP. A Supra-threshold stimulus is required for opening any Na+ channels that have not returned to their resting state; and also to counteract the efflux from the K+ channels that remain open from the repolarization phase.