Molecular mechanism of postconditioning:
It is supported by several authors, that postconditioning the heart is as protective as described in preconditioning experiments. The protective mechanisms seems to be the decreased endothelial dysfunction, reduced oxidative stress and apoptosis and reduction of mitochondrial Ca2+ accumulation. The molecular mechanism induced through postconditioning is similar to the mechanism activated by preconditioning. Ischemic postconditioning induces the reperfusion injury salvage kinase (RISK) pathway, which include the activation of PI3K, Akt and ERK 1/2. Tsang et al. proved in isolated rat heart, that postconditioning is cardioprotective by activating PI3K-Akt pathway, in the initial period of reperfusion. Akt is a serine/threonine kinase, which targeted and phosphorylated by PI3K, and translocated from cytosol to plasma membrane, then phosphorylated by 3-phosphoinositide-dependent protein kinase-1 (PDK1). Darling et al. proved that ERK1/2 is also important for the cardioprotective effect of ischemic postconditioing. They demonstrated that the cardioprotection by postconditioning is abolished by administration of the ERK1/2 inhibitor (PD) and PI3K antagonist (LY) is not shows cardioprotection.
Several studies described that PKG is an important mediator of the ischemic postconditioning. Inserte et al. demonstrated that PKG is essential, and its induced by an NO-cGMP dependent manner. They proved that inhibition of NO-cGMP/PKG pathway abrogates the ischemic postconditioning cardioprotective effect. Zhu et al. found that activation of the PI3K-PKB/Akt pathway is important for inducing cardioprotection by postconditioning. Schwartz et al. proved that ischemic postconditioning applied in the early reperfusion, is cardioprotective, and its caused through phosphorylation of ERK1/2, Akt and p70S6K. Under stress conditions, the mPTP is opening and allows molecules smaller than 1500Da pass into the matrix. This event causes matrix Ca2+ accumulation, oxidative stress and alkalinization. In postconditioning, mPTP has a crucial role in cardioprotection. Argaud et al. demonstarted that postconditioning changes the mPTP and contribute to induced cardioprotection. They reported in rabbit heart, that postconditioning is significantly reduced infarct size, after 72 hours of reperfusion, and mPTP has a major role in it. The role of eNOS in postconditioning is poorly understood. Pong et al. found that in eNOS knockout mice, that in reperfusion the microvascular blood flow is required an activation of eNOS. In addition, eNOS produced NO is an activator of pro-survival kinases, and eNOS activators are achieve cardipoprotection in ischemic and pharmacological postconditioning.
ROS is increased in the early phase of reperfusion, thus contribute to development reperfusion injury. Tsutsumi et al. found both ischemic and pharmacological postconditioning, that a ROS scavenger, MPG is able to decrease the cardioprotective effects of postconditioning. It is suggesting that ROS are also important for the induction the signaling pathway which contribute to cardioprotection through postconditioning. He-Ying Sun et al. described that postconditioning is cardioprotective through a molecular mechanism, which results attenuation of cardiomyocyte loss via reduces ROS and Ca2+ production. (fig. 8)
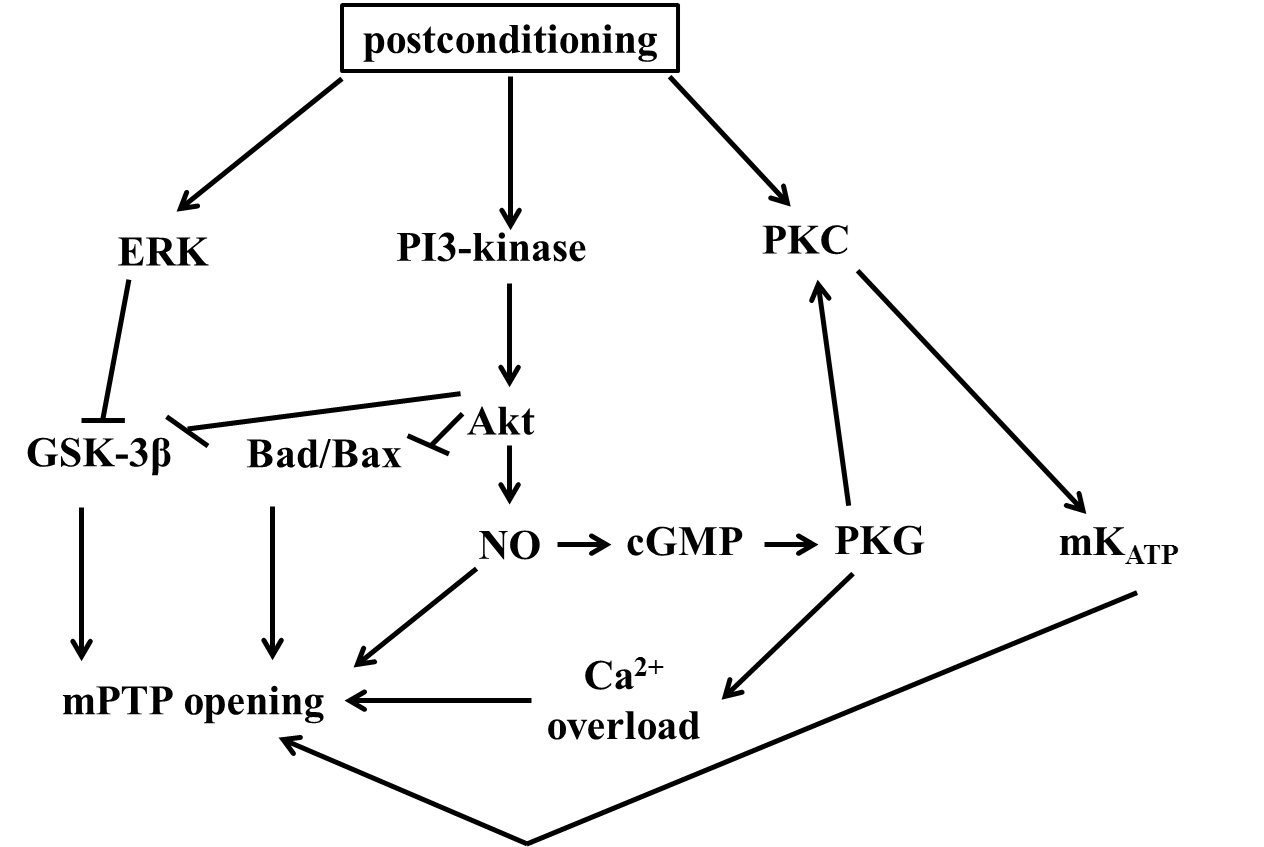
Figure 8. The mechanisms of postconditioning