Coronary occlusion and global ischemia (in vivo, and ex vivo)
Induction of global ischemia (ex vivo)
Animals subjected to ischemic- and reperfusion-associated insults to heart function may be monitored for cardiac functional variables, including heart rate (HR), coronary flow (CF), aortic flow (AF), left ventricular developed pressure (LVDP), left ventricular end-diastolic pressure and (LVEDP). These time-course and drug dosage-related profiles of these endpoints functional clearly show functional status of the heart; and also reveal significant differences between the drug treated and untreated groups or wild type and genetically manipulated mice (such as transgenic and gene knockout animals). Knockout (KO) mice are particularly advantageous models for investigation of signal transduction mechanisms - since lack of important genes may significantly disrupt the cardiac functions, allowing increasingly precise elucidation of pathomechanisms. Experiments which evaluate cardiac functions by the above method are particularly advantageous in terms of the cost-effectiveness of a particular line of investigation, since they may be used as a guide to design more complex experimental protocols – thus sparing expense in design and accomplishment of analysis of molecular-biological features of disease and interventions (Kaga 2005).
In addition to measurement of basic cardiac functions (HR, CF, AF, LVDP and LVEDP), isolated working heart methods allow assessment of ischemia or reperfusion-induced ventricular fibrillation (VF), ventricular tachycardia (VT) and infarct size. Related tests may be conducted for cardiac apoptosis, protein and RNA expression. Induction of ischemia for this kind of experiment may be induced by occlusive clamping of the atrial inflow and aortic outflow lines (global ischemia); or by a suture at a point close to the origin of the coronary arteries (regional ischemia). Reperfusion in this setup is initiated by unclamping the atrial inflow and aortic outflow lines and the coronary arteries. Epicardial ECG readouts may be recorded throughout the experimental period by two silver electrodes attached directly to the heart. One of the silver electrodes contacts directly to heart and another one directly to the aortic cannula. The ECGs can be analyzed to determine the presence or absence of ischemia and reperfusion-induced ventricular arrhythmias. Hearts are considered to be in VF if an irregular undulating baseline is apparent on the ECG. If VT and VF are developed and the sinus rhythm dos not spontaneously return, hearts can be electrically defibrillated by a defibrillator using two silver electrodes and 15 V square-wave pulse of 1 ms duration and reperfused, and then cardiac function can be detected, and biopsies can be taken for molecular and cellular biology studies.
Surgical procedures using the left anterior descending coronary artery (LAD) occlusion, and preconditioning of the heart
Under in vivo conditions, mice, rats, guinea pigs, and rabbits are generally anesthetized with ketamine HCl (100 mg/kg i.p.) and xylazine (10 mg/kg i.p.). Cefazolin (25 mg/kg, i.p.). This aforementioned regimen should be administered as preoperative conditioning workup. After tracheotomy and initiation of ventilation (ambient room air conditions), the heart is exposed through a left lateral thoracotomy (4th intercostal space), a 6-0 polypropylene suture is then passed with tapered needle under the left anterior descending coronary artery (LAD) just below the tip of the left auricle, and a non-traumatic occluder is applied to the artery.
Hearts to be used in experimental procedures, may be treated in ways that can augment native endogenous cardioprotective mechanisms. These procedures, known as “preconditioning” (PC), are accomplished by subjecting the heart to a short duration of temporary regional ischemia following by a period of reperfusion which may be repeated multiple times. Myocardial ischemia is produced by longer periods of LAD occlusion. Preconditioning may be used both in and in vivo model and also in isolated working hearts in a Langendorff apparatus. In rats not subjected to PC, the animals undergo LAD ligation after opening the chest without a PC procedure. After completion of all surgical protocols, the chest wall is generally re-closed and the animal is given buprenorphine (0.1 mg/kg s.c.), followed by weaning from the respirator, the rats should be placed on a heating pad while recovering from anesthesia (Sasaki et al., 2001).
Determination of tissue carbon monoxide (CO) content
CO is produced as a tissue metabolite by degradation of heme by heme oxygenases, acts as a powerful cytoprotective signaling molecule (Haines 2012). Tissue content of this molecule may be measured using gas chromatography can be used as described by (Cook 1995). Briefly, tissues are homogenized in 4 volumes of 0.1 M phosphate buffer (pH 7.4). The homogenates are centrifuged at 4°C for 15 min at 12,800 × g and the supernatant fractions are used for the determination of tissue CO levels. The reaction mixtures used for this assay contain the following components: 150 μl of supernatant, 60 μl of NADPH (4.5 mM), and 50 μl of 3.5/0.35 mM methemalbumin, and for blank samples 60 μl phosphate buffer should be used instead of NADPH. The samples are preincubated at 37°C for 5 minutes; then, the headspace is purged and the incubation is continued for 1 hour in the dark at 37°C. The reaction is stopped by placing the samples on ice and the headspace gas is analyzed. One thousand microliters of the headspace gas from each vial is injected into the gas chromatograph using a gas-tight syringe in argon gas flow with a speed of 20 ml/min. Analyses for CO content are conducted during the next 90 seconds on a 240 cm stainless-steel column with a 0.3 cm inner diameter. The individual value is expressed in mV; following which, the peak’s area is integrated and expressed in arbitrary units. The column is packed with Chromosorb 80/100 mesh (10% Carbowax 20 M, 3.5% KOH) and maintained at 120°C. The temperature of the injector should be controlled and kept at 150°C (Bak 2002).
Heme oxygenase (HO) Activity Assay
Heme oxygenases (HO) are enzymes expressed ubiquitously by most tissues and constitute major antioxidant defense mechanisms. Three isoforms exist: HO-1, HO-2 and HO-3, each of which metabolizes heme deposits that accumulate in tissues due to red blood cell turnover. Two products of this degradation, carbon monoxide (CO) and bilirubin – have potent capacity for reducing oxidative stress and for counteracting its effects. The Tosaki Laboratory has focused particular attention on HO-1, the inducible form of the enzyme, on which this laboratory has developed a number of very powerful therapeutic approaches which make use of a phytochemical inducer of HO-1 which has recently proven highly successful in abatement of osteoarthritis symptoms in a phase 1 human clinical trial (Mahmoud 2014).
The methodology used for quantitating HO protein expression by a particular tissue are described as follows: Biopsies from various tissues are homogenized in 10 mL of 200 mM phosphate buffer, and the homogenate is centrifuged at 19,000g 4°C for 10 minutes. The supernatant is removed and recentrifuged at 100,000g 4°C for 60 minutes, and the precipitated fraction is suspended in 2 mL of 100 mM potassium phosphate buffer. Biliverdin reductase is crudely purified by the technique of Tenhunen et al (1970). HO activity can be assayed as described by Yoshida et al (1974). Reaction mixtures consisted of 100 µM potassium phosphate (pH 7.4), 15 nM hemin, 300 µM bovine serum albumin, 1 mg biliverdin reductase, and 1 to 2 mg microsomal fraction of the tissue (final volume of 2 mL). The reaction is allowed to proceed for 1 hour at 37°C in the dark in a shaking water bath and is stopped by placing the test tube on ice. Incubation mixtures are scanned using a scanning spectrophotometer, and the amount of bilirubin is calculated as the difference between absorbance at 464 and 530 nm. Protein content is determined with Folin-phenol reagent according to Lowry et al (1951) in the microsomal fraction.
Inducible nitric oxide synthetase (INOS) activity
Nitric oxide (NO) is a reactive oxygen species, which in high concentrations may damage tissue by its contribution to several important consequences of oxidative stress, in particular, lipid peroxidation. At lower levels, NO is a critically important signalling molecule that is responsible for a wide range of normal and pathological physiologic activities, particularly regulation of vascular tone. Its tissue levels are regulated by nitric oxide synthetase (NOS), the enzyme that L-arginine into citrullin and NO. A schematic representation of this conversion is shown below:
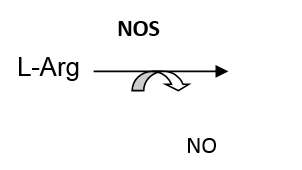 Citrullin L-Arg : citrullin = 1:1 and NO : citrullin = 1: 1
Citrullin L-Arg : citrullin = 1:1 and NO : citrullin = 1: 1
NOS isoforms: Several NOS variants are present in vertebrate tissues as described below.
INOS: Inducable nitric oxide synthase (activity is [Ca2+]-independent)
cNOS: Constituitive nitric oxide synthase (activity is [Ca2+]-dependent)
nNOS: Neuron-associated NOS
eNOS: Endothelium-associated NOS
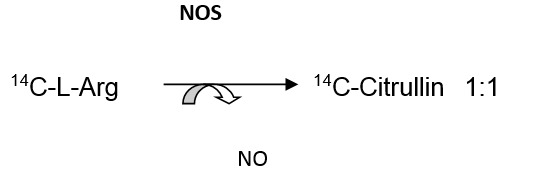
Assessment of tissue NO content is typically accomplished indirectly, through measurement of related metabolites. Shown below are approaches to NO production that make use of metabolites such as citrullin:
Basic citrullin production relationship:
Electron spin resonance (ESR) for the measurement of oxygen reactive species
Spin trap studies may be performed by infusing a spin trap, e.g., PBN and DMPO, into the entire body or organs of an experimental animal. To prevent light-induced degradation of spin traps, the infusion syringe is covered. Additionally, spin-adduct decay should be prevented. This may be accomplished by freezing the effluent in liquid nitrogen as it flows from tissues with an effluent sampling time of 30 sec. ESR spectra may be recorded in a flat quartz cell with a spectrometer operating at X band (9.3 MHz) with a 100-kHz modulation frequency. The microwave power is maintained at 10 mW to avoid saturation. Scans are traced with 0.2 mT of modulation amplitude with 2 min of scan time and 300 ms of response time. Hyperfine coupling constants are measured directly from the field scan by using Mn2+ as a marker for calibration (Pataki 2002).
ESR for the measurement of nitric oxide (NO)
The spin-trap diethyl-dithio-carbamate (DETC), FeSO4, and sodium-citrate are slowly administered intravenously into the femoral vein of rodents. DETC dissolved in distilled water is injected separately form FeSO4 and sodium-citrate in 0.5 ml volume to avoid precipitation of Fe2+-(DETC)3. FeSO4 and sodium-citrate are dissolved in distilled water, pH is set to 7.4 with NaOH, and brought to 1 ml volume before injection. Five min after DETC, FeSO4 and citrate treatment, tissues (e.g., myocardium) are isolated and perfused to washout blood, and 100 mg of the biopsy are placed into quartz tubes, and frozen in liquid nitrogen until assayed for ESR spectra of NO-Fe2+-(DETC)2 complex. In isolated and perfused tissues, a side-arm in the perfusion line allowed direct infusion of the FeSO4 and DETC, NO-Fe2+-(DETC)2-adduct is registered. Fe2+-(DETC)2 complex has high affinity to nitric oxide forming the NO-Fe2+-(DETC)2 adduct. The specific triplet signal-adduct of the NO-Fe2+-(DETC)2 complex is superimposed on the dominant background spectra of Cu2+-(DETC)2. Background spectra of Cu2+-(DETC)2 complex can be determined in tissues treated with DETC only (). The detection limit of NO by this ESR method is 0.05 nM (Mulsch et al., 1992). ESR spectra can be recorded with ESR spectrometer operating at X band with 100 kHz modulation frequency at a temperature of 160 oK, using 10 mW microwave power to avoid saturation. Scans are traced with 2.85 G modulation amplitude, 150 G sweep width, and 3356 G central field as described (Mulsch 1992; Zweier 1995, Ferdinandy 1996).
Glutathione oxidation
Another common and widely accepted method of detecting free radical production in exercise is by measuring the levels of glutathione (L-γ-glutamyl-cysteinyl-glycine) that is intimately involved in some of the body's most important antioxidant defense systems. Free radicals and oxidant species, such as H2O2, can oxidize reduced glutathione (GSH) to form an (oxidized) glutathione disulfide dimer (GSSG). This reaction is catalyzed by glutathione peroxidase. Since, glutathione reductase can split the dimmer and reduce both monomers back to GSH, this forms an important antioxidant enzyme system for removing H2O2 and other cellular oxidants. GSH levels have typically been measured in blood, liver, and muscle tissues and have been shown to decrease after exhaustive exercise, with concomitant increases in GSSG levels (Sachdev 2008).
Analysis of Atherosclerotic Lesions. Quantification of fatty streaks is performed with Sudan III stain. Thoracic arteries are harvested, dissected free of excess connective tissue and fat, rinsed with modified Krebs-Henseleit buffer, and fixed in 10 % (vol/vol) buffered formalin. Carotid arteries are then opened longitudinally, and exposed to 5 mg/mL Sudan III in 70 % (vol/vol) isopropanol for 15 minutes in a water bath at 37 °C, and the stain is differentiated with several rinses of 70 % isopropanol. The arteries are then scanned, and the atherosclerotic plaque is determined (Juhasz 2013, Kertesz 2013).
Cytochrome c Oxidase (COX) Activity. The activity of cytochrome c oxidase in animal (mice, rat, rabbit) myocardium is measured using a colorimetric assay kit for oxidation of cytochrome c by this enzyme (Sigma-Aldrich, St. Louis, Missouri, U.S.A.). Briefly, mitochondria is isolated from freshly harvested heart muscle using the MITOISO1 kit (Sigma-Aldrich, St. Louis) should be treated with dithiothreitol to reduce cytochrome c, follows by COX-mediated reoxidation of the molecule. COX activity at room temperature (~22 °C) in each sample is measured as a decrease in absorbance of ferrocytochrome c, at an absorbance wavelength of 550 nm (UV Helios Alpha S2 spectrophotometer) in its conversion from a reduced to oxidized state. The COX activity in any particular sample is reported as (-ΔA550/min) (Juhasz 2013, Kertesz 2013).