Molecular mechanism of preconditioning:
Preconditioning is a potent manouver for reduce the damage caused by ischaemia-reperfusion injury, and reduced the infarct size. The molecular mechanism produced by preconditioning is not fully described yet. Numerous studies demonstrated the beneficial signaling pathways induced by ischemic preconditioning and lead to cardioprotection. Several serine/threonine kinases, termed survival kinases, including protein kinase C (PKC), phosphatidylinositol 3-kinase (PI3K), Akt, p38 mitogen-activated protein kinase (MAPK), p42/44 MAPK/ERK, Src family receptor tyrosine kinases and the JAK/STAT pathway are essential for regulate cell survival. PKCs are lipid sensitive enzymes, activated by growth factor receptors that stimulate phospholipase C (PLC), which hydrolyzes phosphatidil-inositol 4,5-bisphosphate (PIP2) into inositol 1,4,5-triphosphate (IP3) and dyacilglicerol (DAG). The produced DAG molecule is membrane-bound and able to activate PKCs. PKCs are catalyze phosphorilation reactions to their targets, in the ser/threonine and tyrosine residues. Cardioprotective agonists are able to activate PLC, which thereby observed in an increased activation of PKC. PKC seems to be a central mediator of ischemic preconditioning, which demonstrated first by Armstrong et al., with calphostin, a PKC specific inhibitor. Calphostin administration decreased the protective effect of preconditioning. There are several controversial studies about the role of PKC in cardioprotection. Vogt et al. showed that pharmacologically inhibition of PKC led to cardioprotective effects. Ping et al. demonstrated that from the existing 11 isoforms of PKC, only two isoforms, the PKCη and PKCε are undergo trasnlocation in preconditioning, led to the discovering, that PKC activation by preconditioning is isoform selective. Evidence shows that also protein kinase A (PKA) and cAMP are expressed higher in ischemic preconditioning, which thereby seems to suggest a role in it. Sanada et al. suggest that PKA cardioprotective effect is seems to related to the activation of p38MAPK. p38MAPK is a member of MAP kinases, which activation is more sensitive to stress signals, such as oxidative stress, heat shock and UV radiation. Several studies demonstrated that inhibition of p38MAPKs decreased the efficacy of cardioprotection. Mocanu et al. described that a p38 MPK inhibitor, SB203580, is cause decrease in cardioprotection, only that case when the inhibitor administration was during sustained ischemic period. (The p38 MAPK inhibitor, SB203580, abrogates ischaemic preconditioning in rat heart but timing of administration is critical) Another family of MAPK is seems to be essential for the develop the effect of protection, the p42/44MAPK. It is becoming clear that both PI3K/Akt and p42/44MAPK are highly phosphorylated in ischemic preconditioning at he time at reperfusion, and this phosphorylation seems to be important for developing the protective effect. Wynne et al. demonstrated that administration of pioglitazone is associated with the increase of protection against infarction, in isolated rat heart. They suggest that the protective effect is caused by PI3K activation at pioglitazone administration and both activation of PI3K and p42/44MAPK is required at reperfusion. (Pioglitazone Mimics Preconditioning in the Isolated Perfused Rat Heart) The role of extracellular signal-regulated kinase (ERK1 and ERK2) is also seems to be important. The ERK cascade is essential for cellular processes, such as proliferation, differentiation, through activation of various antiapoptotic mechanisms. The role of ERKs in preconditioning is controversial. Several studies reported a role for ERKs in cardioprotection, (ROS-mediated ERK activation in delayed protection from anoxic preconditioning in neonatal rat cardiomyocytes) however, others are demonstared the opposite results. JAK/STAT pathway seems to essential for the protective effect of ischemic preconditioning. Xuen et al. have found, that ischemic preconditioning induces highly activation of JAK1, JAK2, STAT1 and STAT3, which are contribute to cardioprotection. Adenosine is also play a crucial role in the mechanism of preconditioning. The known adenosine receptors, are A1, A2A, A2B and A3. A1ARs and A2ARs are expressed only in the adult myocardium Several studies suggest that there is an important role for the A3 receptor of adenosine too. Pharmagological agonists, CCPA, an A1 agonist and IB-MECA, an A3 agonist, administration is imitate ischemic preconditioning. Zhao et al. have proved that preconditioning protection by administration of CCPA agonist is because of the increased nitric oxide synthesis. This cardioprotective effect could terminate by an iNOS inhibitor agent. It is also found that A1AR agonism operate on several kinases, such as PKC and MAPKs, and A1AR antagonism is seems to suppress the translocation of PKC, thereby decreases the effect of cardioprotection. In addition, opening the KATP channels also seems to contribute to the mediation of the AR induced cardioprotection. KATP channels contribute two subunits, a small KIR, an a large sulphonilurea receptors (SURs). Evidence shows that there are two types of KATP channels in a cell, the sarcolemmal and the mitochondrial types, which placed in the inner membrane of mitochodrium. The opening of sarcolemmal ATP dependent K+ channel is induced by ischemia and hypoxia, and because of the potassium influx, led to a shorten action potential duration, by promote repolarisation. This causes inhibition of L-type Ca2+ channels and hinder Ca2+ overload. Mitochondrial KATP channel is not a trigger for preconditioning but essential for the preconditioning induced signalling pathway. Opening mitochondrial potassium channels are PKG dependent. PKCε is the transmitting protein, between cytosolic PKG and mitochondrial KATP channel. PKCε is essential for achieve the cardioprotective event, and Dorn et al. found that another isoform, PKCδ is responsible for blocking the protection. From the aspect of preconditioning the important mechanism is the redox coupling of mitochondrial KATP channel and PKC activation. Vanden Hoek et al. described that 5-hydroxydecanoate (5-HD), which a KATP channel blocker, and a PKC inhibitor, Go-6976, both decreased the preconditioning caused attenuation of oxidative stress, This result suggest, that this mechanism is mediated the antioxidant system. Thereby reactive oxygen species are important for the signaling pathway of preconditioning. Vanden Hoek et al. reported that administration of exogenous ROS, such as H2O2 is responsible for preconditioning cardioprotection. Furthermore, they proved that involvement in preconditioning, mitochondrial KATP channel in increased ROS production, thus cardioprotection, is necessary. However, the exact relationship between mitochondrial KATP channel and increased ROS porduction is poorly understood. It is suggested, that the PKG dependent opening of mitochondrial KATP channel, cause K+ influx to the mitochondria matrix, and an electrogenic H+ efflux. This mitochondria matrix alkalinization activates complex I and/or III, and thus generate highly amount of ROS, including superoxide, H2O2 and hydroxyl radical. Furthermore, the inner membrane localized connexin 43, seems to play a crucial role in the generating of ROS also. It’s proved that on connexin 43 deficient mice preconditioning is not represent cardioprotection. It is also hypothesized that openeing mitochondrial permeability transition pore (mPTP) is also contribute to ROS production. Hausenloy et al. found that transiest openinsg of mPTP is responsible for increased production of ROS, and seems to be cardioprotective in both ischemich and pharmacological preconditioning. The role of NO in preconditioning is controversial. Woolfson et al found that administration of L-NAME, a NOS inhibitor, has reduced infarct size in preconditioning induced cardioprotection. Controversely, Bilinska et al. described that adding an NO donor, specifically contribute to the cardioprotection of preconditioning. Several authors suggested, that bradykinin is a major mediator of preconditioning. Bradykinin is a potent vasodilator peptide in the cardiovascular system. Schoelkens et al. reported for the first time, that administration of exogenous bradykinin is cardioprotective and seems to operate with preconditioning. It is becoming increasingly clear, that in precondetioned heart, the responsible for protection is the pathway activating PKC by PI3K-Akt-eNOS-cGMP-PKG pathway, which is permit to open ATP dependent mitochondrial K+ channel (mKATP). The opening of this channel cause an increased ROS level in the cytoplasm, which seems to be important for activating PKCs and explicate protection. (Survival kinases in ischemic preconditioning and postconditioning) (fig. 4)
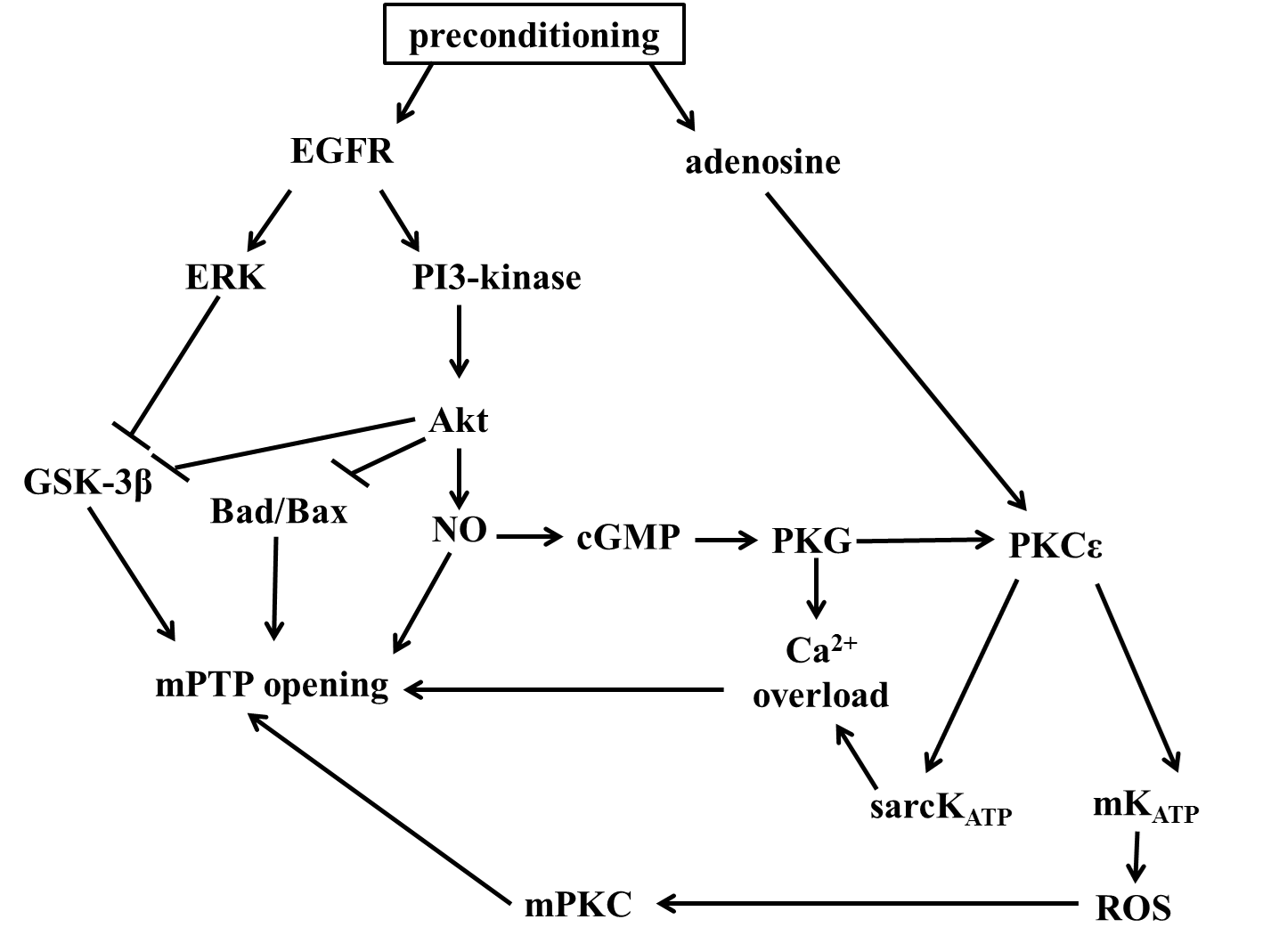
Figure 7. The mechanisms of preconditioning